


Critical challenges identified in 2023 US FDA audits of sterile facilities encompass environmental monitoring, cleaning, disinfection, microbiological control, and process validation. Manufacturers are urged to prioritize and enhance protocols in these areas, which is imperative for maintaining a commitment to delivering safe, high-quality drug products aligned with regulatory standards.

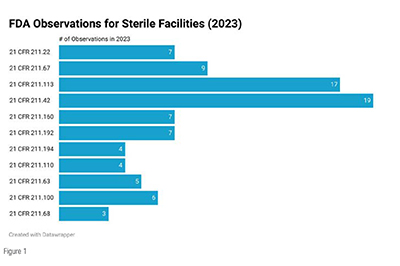
The sterile pharmaceutical market is expected to expand significantly in the coming years due to the increasing demand for biological drugs, Advanced Therapy Medicinal Products (ATMPs)/Cell and gene Therapies (CGTs), and generic sterile injectable. The global sterile manufacturing market is projected to grow by more than 50% by 2028. Developing advanced aseptic manufacturing techniques has improved sterile injectable drugs' quality, safety, and shelf-life, contributing to market growth. However, challenges such as the shortage of sterile-manufacturing talent and regulatory GMP compliance may hinder the rapid expansion of sterile capacity. In sterile manufacturing, GMPs are essential for preventing contamination and ensuring the safety and efficacy of the final products. Sterile Drug Products Produced by Aseptic Processing- Current Good Manufacturing Practice Guidance for Industry provides the US FDA’s views on aspects the manufacturers must meet when manufacturing sterile drugs and biological products using aseptic processing for US FDA-regulated products/processes. The article investigates GMP violations observed, particularly during FDA inspections of sterile facilities in 2023. The 2023 dataset was sourced from the US FDA compliance dashboards. Citation data is only available for inspections where all area classifications are finalized. Citations for manually prepared 483s are also excluded. Figure 1 presents the CFR section that generated three or more observations for 2023.
The top three sterile facility audit observations from the US FDA in 2023 were related to 21 Code of Federal Regulations (CFR) 211 sections 42, 113, and 67. At the top is Section 211.42 of the CFR Title 21, addressing design and construction features. This critical aspect ensures that sterile environments are meticulously crafted to meet stringent specifications, safeguarding the integrity of pharmaceutical production. The second most observed stems from Section 211.113, which focuses on the control of microbiological contamination. Lastly, Section 211.67, centered on equipment cleaning and maintenance, is vital in maintaining sterile conditions. The meticulous upkeep of equipment is paramount to prevent cross-contamination and maintain the hygienic standards indispensable to sterile facility operations. These audit observations serve as a clarion call for pharmaceutical manufacturers to prioritize these critical regulatory areas.
Delving into the infractions concerning design and construction features (Section 211.42), a noteworthy revelation emerged: a staggering 42% of violations were directly linked to inadequacies in the monitoring system for environmental conditions. Consequently, it becomes imperative to meticulously integrate justified methodologies for monitoring environmental conditions. Additionally, 26% were attributed to the cleaning and disinfecting of the room and equipment. Developing robust processes for cleaning and disinfecting the room and equipment is therefore essential. Activities must take place within precisely designated, appropriately sized areas. Distinct areas and controls should be established as needed to forestall any potential cross-contamination or mix-ups during the course of manufacturing operations. Using isolators where appropriate and regularly monitoring air cleanliness in critical areas are essential. Additionally, cleanroom qualifications should include an assessment of air quality under dynamic conditions, with personnel present and operations ongoing. By meticulously integrating justified methodologies for monitoring environmental conditions and developing robust processes for cleaning and disinfecting, manufacturers can address FDA concerns and ensure ongoing compliance with cGMP’s.
To manage microbiological contamination effectively, it is essential to implement suitable written procedures. These procedures should be crafted to prevent the presence of objectionable microorganisms in drug products. This includes rationalized validation study protocols for all aseptic and sterilization processes that are involved. A control strategy should be in place to prevent potential microbial contamination in all operating conditions and the treatment of materials. This includes implementing measures such as regular cleaning, disinfection, and maintaining a high level of hygiene in every aspect of the manufacture of drug products. Automation and barrier technology, adherence to first-air principles, and good aseptic technique/behavior are vital parts of a prevention strategy. It's crucial to highlight that nearly one-third of observations under Section 211.113, precisely 29%, of the observations made were directly linked to the absence of comprehensive process validation studies. This indicates a significant gap in ensuring the robustness and reliability of the manufacturing process. Process validation studies confirm that a given process consistently produces a product meeting its predetermined specifications and attributes. Addressing process validation issues requires a meticulous and thorough data-driven manufacturing science-focused approach in development (Stage 1- QbD), qualification (Stage 2 PPQ), and monitoring (Stage 3- CPV).
Ensuring the integrity and safety of drug products necessitates a meticulous approach to managing equipment and utensils. The tools undergo regular cleaning, maintenance, and sanitization. The goal is to prevent contamination and malfunctions that could compromise the drug product's safety, identity, strength, quality, or purity. To uphold these standards, written procedures must be established and adhered to for the systematic cleaning and maintenance of equipment, including utensils, employed in manufacturing, processing, packing, or holding. Section 211.67(a) observations, which constituted 66%, are specific to not performing the equipment and utensils cleaning and sanitization at appropriate intervals to prevent contamination. The remaining observations in this section pertained to the absence or inadequacy of procedures for equipment cleaning and maintenance. Notably, within Section 211.42, a significant number of observations were linked to the cleaning and disinfection of equipment and rooms, emphasizing the scrutiny placed on aseptic area cleaning practices as one of the most frequently observed aspects. This underscores the critical need for pharmaceutical facilities to prioritize and enhance protocols related to equipment cleaning, sanitization intervals, and maintenance procedures to align with regulatory expectations.
The 2023 observations from US FDA inspections shed light on critical areas requiring attention within sterile facilities. The prominence of Section 211.42, emphasizing design and construction, Section 211.113, focusing on microbiological contamination control, and Section 211.67, highlighting equipment cleaning and maintenance, reflects the multifaceted challenges faced by the pharmaceutical sector. The identified gap in process validation studies, constituting 29% of Section 211.113 observations, underscores the imperative for a comprehensive, data-driven approach throughout the manufacturing process life cycle. Furthermore, the emphasis on equipment cleaning, sanitization intervals, and the scrutiny of aseptic area cleaning practices signals a clear mandate for pharmaceutical facilities to prioritize and enhance protocols, aligning with regulatory expectations. In navigating the evolving landscape of sterile pharmaceuticals, addressing these observations is crucial for sustaining the industry's commitment to delivering safe, high-quality products that meet regulatory standards and ensure patient well-being.
References:
1. https://www.mckinsey.com/industries/life-sciences/our-insights/how-sterile-pharma-manufacturers-can-grow-capacity-without-capital-investment
2. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/sterile-drug-products-produced-aseptic-processing-current-good-manufacturing-practice
3. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=211.42
4. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=211.113
5. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=211.67

Ajay Pazhayattil is a management consultant and an industrial pharmacist with pharmaceutical operational experience in the industry's sterile, solid oral, and API sectors. He has been in leadership roles with North American brand, generic, and CDMO organizations before establishing cGMP World, an organization helping pharmaceutical firms maintain regulatory compliance. He has successfully remediated warning letter scenarios and developed regulatory acceptable unique compliance solutions. He can be contacted through LinkedIn.

Marzena Ingram is an independent senior pharmaceutical consultant with extensive quality and technical operations experience in solid-dose and active pharmaceutical ingredient manufacturing operations. She is responsible for delivering compliant solutions for clients addressing FDA warning letter scenarios. Ingram has spearheaded compliance programs meeting global regulatory requirements. She is the vice president of ISPE Canada. She can be contacted through LinkedIn.

")





